概要
LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) は、古典分子動力学に関するソフトウェアです。LAMMPS は、ソリッドマテリアル (金属や半導体)、ソフトマテリアル (生体高分子やポリマー)、および粗視化モデルシステムやメゾスコピックモデルシステムに使用されます。
公式サイトを表示するには、こちらをクリックします。
3d Lennard-Jones 融解の例
前提条件
- クラスターの作成時に LAMMPS ソフトウェアパッケージをインストールします。
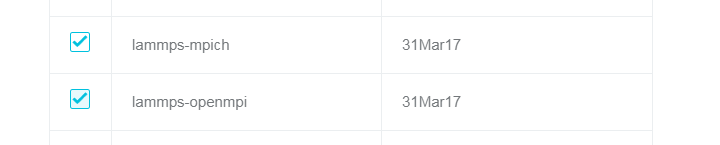
- ソフトウェアが依存する MPI ライブラリを選択します。
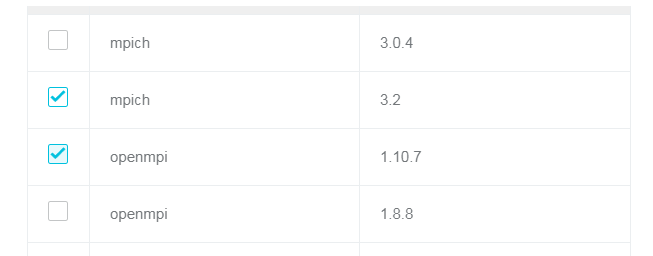
手順
モジュール avail を実行して、LAMMPS ソフトウェアがインストールされているかどうかを確認します。
$ export MODULEPATH=/opt/ehpcmodulefiles/$ module avail------------------------------ /opt/ehpcmodulefiles -------------------------------------lammps/31Mar17-mpich lammps/31Mar17-openmpi mpich/3.2.2 openmpi/1.10.7
モジュール load を実行して、LAMMPS をロードします。
$ module load lammps/31Mar17-mpich$ module load mpich$ which lmp/opt/lammps/31Mar17-mpich/lmp
後述するジョブ送信方法の詳細は、サンプルディレクトリにアクセスしてください。
- コマンドラインを使用してジョブを送信
$ srun --mpi=pmi2 -N 2 -n 4 lmp -in in.intel.ljLAMMPS (31 Mar 2017)Lattice spacing in x,y,z = 1.6796 1.6796 1.6796Created orthogonal box = (0 0 0) to (134.368 67.1838 67.1838)2 by 1 by 2 MPI processor gridCreated 512000 atoms... ...
- ジョブ形式でジョブを送信
$ cat job.sh # Job content#!/usr/bin/env bashmpirun lmp -in ./in.intel.lj$ sbatch -N 2 -n 4 ./job.sh # Submit jobSubmitted batch job 235$ squeue # View jobJOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)235 comp job.sh user R 0:03 2 s[02-03]
- リソースの割り当て後にジョブを送信
$ salloc -N 2 mpirun -n 4 lmp -in in.intel.ljsalloc: Granted job allocation 236salloc: Waiting for resource configurationsalloc: Nodes s[02-03] are ready for jobLAMMPS (31 Mar 2017)
- PBS ジョブの送信 (GPU アクセラレーション)
$ cat > lammps_single_node.pbs#!/bin/sh#PBS -l ncpus=28,mem=12gb#PBS -l walltime=00:10:00#PBS -o lammps_pbs.log#PBS -j oecd /opt/lammps/31Mar17-openmpi/src cd / opt / lammps / 31Mar17-openmpi / src/opt/openmpi/bin/mpirun -np 28 /opt/lammps/31Mar17-openmpi/bin/lmp_mpi -sf gpu -pk gpu 2 -in ./in.intel.lj -v m 0.1$ qsub lammps_single_node.pbs