概要

GROMACS (GROningen MAchine for Chemical Simulations) は、数百万の粒子を持つ系にニュートンの運動方程式を適用する、分子動力学シミュレーションに使用される一般的なソフトウェアパッケージです。タンパク質、脂質、核酸など、多様で複雑な結合性相互作用を有する生体物質に主に使用されます。非結合性相互作用などの典型的なコンピューティングシミュレーションの効率が高いため、多くの研究者が非生物学的な (高分子化合物などの) 研究にも使用しています。
現代の分子動力学法で使用される一般的なアルゴリズムをすべてサポートしており、そのコードは世界中の開発者によってメンテナンスされています。詳細については公式ウェブサイトをご覧ください。
前提事項
GROMACS ソフトウェアパッケージをクラスターの作成前にインストールしていること。
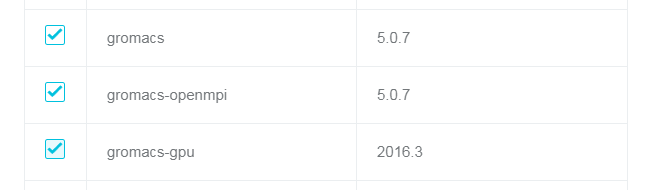
注意:
gromacs-gpu の例を実行するには、クラスター作成の際に、コンピューティングノードに GPU を搭載したインスタンスを選択する必要があります。そうしなければ、例 2 にある通りに gromacs-gpu を実行することはできません。
GROMACS の例を実行する前に、「ジョブの送信」に記述されている前提条件を満足する必要があります。
また、GROMACS ソフトウェアが依存する MPI ライブラリを選択する必要があります。
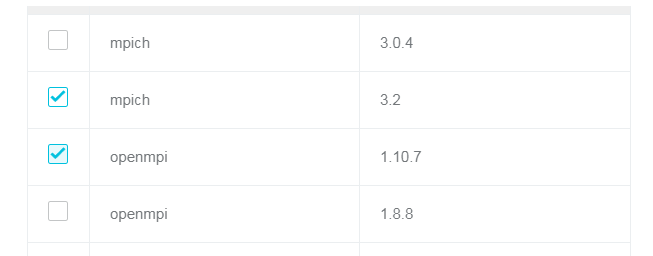
実行例
例 1: 水中リゾチーム
この例では、水槽内の蛋白質 (リゾチーム) とイオンをシミュレートします。詳細は、公式チュートリアルをご覧ください。また、パッケージをダウンロードしてください。
実行手順
シリアル版
$ ./serial_run.shパラレル版
$ ./parallel_run.sh
例 2: 水分子運動
この例では、一定の温度で一定の空間内にある多数の水分子の運動をシミュレートします。GPU インスタンスが必要です。
実行手順
環境変数を設定し、
module availを実行して GROMACS ソフトウェアがインストールされているかどうかを確認します。$ export MODULEPATH=/opt/ehpcmodulefiles/ # The environmental variables needed by the module command$ module avail------------------------------ /opt/ehpcmodulefiles -------------------------------------gromacs-gpu/2016.3 openmpi/1.10.7
module loadを実行して GROMACS と OpenMPI をロードします。$ module load openmpi$ module load gromacs-gpu$ which gmx_mpi/opt/gromacs-gpu/2016.3/bin/gmx_mpi
水分子運動の例をダウンロードします。
現在のディレクトリが現在のユーザーの $HOME パスの下にあるとします。
$ pwd/home/<current_user_name>$ wget http://public-ehs.oss-cn-hangzhou.aliyuncs.com/packages/water_GMX50_bare.tar.gz$ tar xzvf water_GMX50_bare.tar.gz
PBS ジョブを送信して水分子運動の例を実行します。
高性能コンピューティングノードの PBS ジョブスクリプト(> 32 CPU コア、デュアル GPU カード)
$ cat > gromacs_single_node.pbs#!/bin/sh#PBS -l ncpus=32,mem=4gb#PBS -l walltime=00:20:00#PBS -o gromacs_gpu_pbs.log#PBS -j oecd /home/water-cut1.0_GMX50_bare/1536/opt/gromacs-gpu/2016.3/bin/gmx_mpi grompp -f pme.mdp -c conf.gro -p topol.top -o topol_pme.tpr/opt/openmpi/1.10.7/bin/mpirun -np 4 /opt/gromacs-gpu/2016.3/bin/gmx_mpi mdrun -ntomp 8 -resethway -noconfout -nsteps 8000 -v -pin on -nb gpu -gpu_id 0011 -s topol_pme.tpr
低性能ノードの PBS ジョブスクリプト
$ cat > gromacs_single_node.pbs#!/bin/sh#PBS -l ncpus=4,mem=4gb#PBS -l walltime=00:20:00#PBS -o gromacs_gpu_pbs.log#PBS -j oecd /home/water-cut1.0_GMX50_bare/1536/opt/gromacs-gpu/2016.3/bin/gmx_mpi grompp -f pme.mdp -c conf.gro -p topol.top -o topol_pme.tpr/opt/openmpi/1.10.7/bin/mpirun -np 1 /opt/gromacs-gpu/2016.3/bin/gmx_mpi mdrun -ntomp 4 -resethway -noconfout -nsteps 8000 -v -pin on -nb gpu -s topol_pme.tpr
PBS ジョブスクリプトを使ってジョブを送信します。
$ qsub gromacs_single_node.pbs1.iZ2zedptfv8e8dc9c2zt0tZ$ qstatReq'd Req'd ElapJob ID Username Queue Jobname SessID NDS TSK Memory Time S Time--------------- -------- -------- ---------- ------ --- --- ------ ----- - -----1.iZ2zedptfv8e8 mingying workq gromacs_si 20775 1 4 4gb 00:20 R 00:03